Identification de gènes centraux liés au vieillissement : Une étude sur C. elegans
Découvrez comment des gènes centraux identifiés chez C. elegans pourraient révolutionner notre compréhension du vieillissement et ses mécanismes complexes.
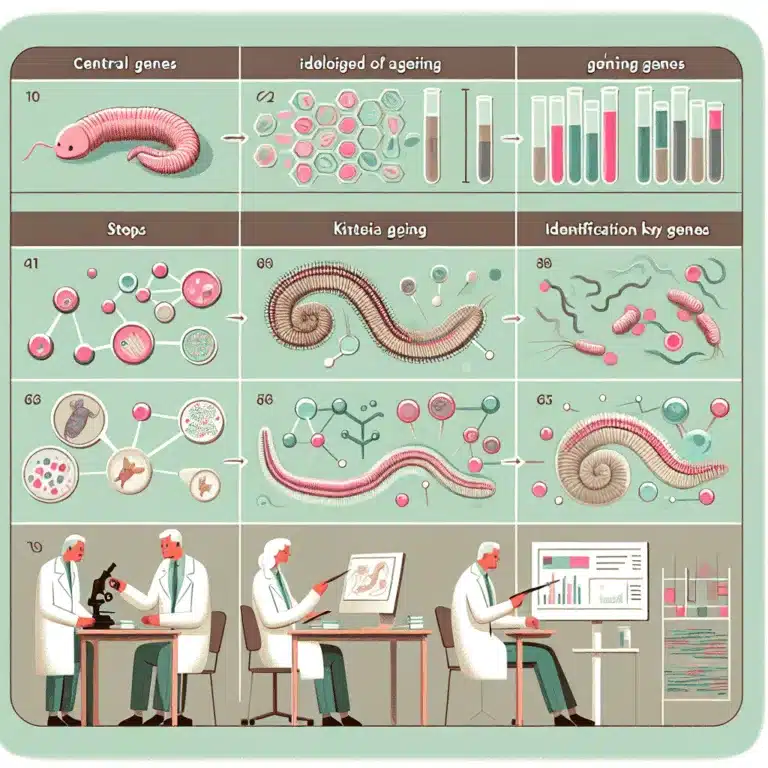
Découvrez comment des gènes centraux identifiés chez C. elegans pourraient révolutionner notre compréhension du vieillissement et ses mécanismes complexes.

Les horloges de vieillissement sont des outils innovants qui peuvent être élaborés à l’aide de techniques d’apprentissage automatique, en utilisant des ensembles de données biologiques complexes provenant de personnes de divers âges. Un algorithme spécifique permet de relier les changements liés à l’âge dans ces données à l’âge chronologique moyen. Lorsqu’on applique cet algorithme à…

La recherche sur le reprogrammation cellulaire vise à induire certains aspects du changement dramatique de l’expression génique et de la fonction cellulaire qui se produisent durant le développement embryonnaire précoce, lorsque les cellules germinales adultes éliminent les modifications épigénétiques caractéristiques de l’âge pour redevenir des cellules souches embryonnaires jeunes. La fonction cellulaire redevient juvénile, et…
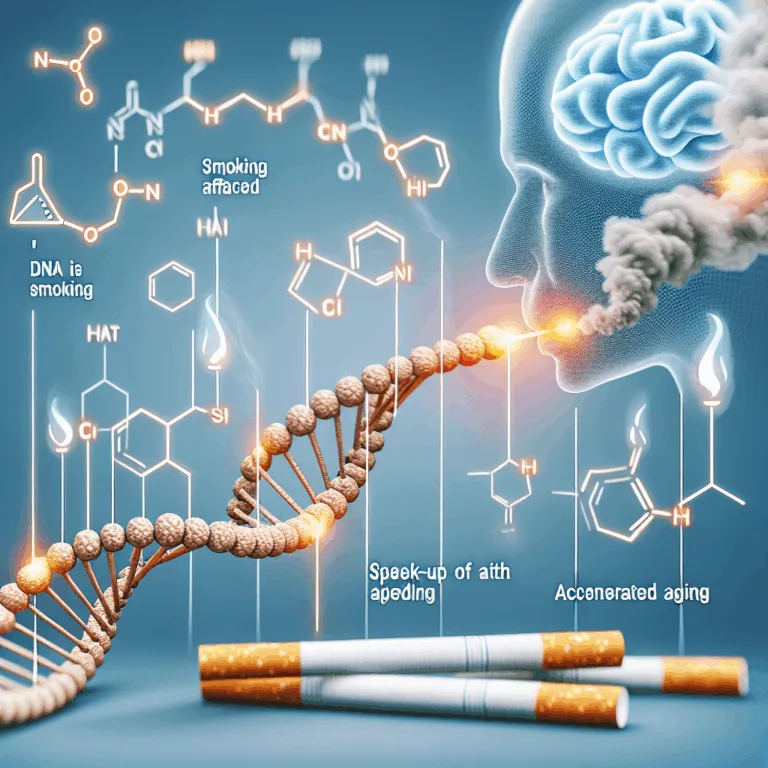
Des chercheurs ont analysé les motifs moléculaires provenant de différents tissus obtenus chez plus de 700 personnes et ont appris que le tabagisme agit comme un accélérateur de vieillissement et implique des changements moléculaires dans des tissus au-delà de ceux directement exposés à la fumée de cigarette. Malgré les campagnes visant à réduire le tabagisme,…

Une étude récente a examiné les différences de potentiel de durée de vie maximale parmi diverses espèces de mammifères. Les chercheurs ont trouvé des associations entre l’expansion de la taille des familles de gènes, le potentiel de durée de vie maximale et la taille relative du cerveau. Ils ont également étudié les caractéristiques génomiques liées…

Des chercheurs publiant dans Aging Cell ont utilisé de grandes bases de données pour découvrir une relation causale entre plusieurs gènes et le risque de mortalité globale, identifiant ainsi un nouveau potentiel cible dans ce processus. Dans leur étude, ils discutent des bases de données génétiques, qui ont été précédemment utilisées pour déterminer les associations…

La structure de l’ADN dans le noyau cellulaire joue un rôle essentiel dans la transcription des gènes et, par conséquent, dans la production d’ARN et de protéines. Ce processus est influencé par la configuration de l’ADN nucléaire, qui peut être modifiée par des facteurs tels que la méthylation et les modifications des protéines histones, affectant…
Cette discussion porte sur la pertinence des changements liés à l’âge dans la régulation épigénétique de l’expression génétique par rapport à la fonction de la mémoire. Le comportement d’une cellule est déterminé par la structure de l’ADN nucléaire, qui détermine quelles régions sont accessibles à la machinerie de transcription responsable de la production de molécules…
L’impact du vieillissement sur les changements transcriptionnels dans les cellules est un domaine de recherche important. En examinant le transcriptome des cellules musculaires des rats âgés par rapport à ceux des jeunes et en tenant compte des interventions comme la restriction calorique, les chercheurs ont pu mieux comprendre les mécanismes sous-jacents à la sarcopénie, qui…
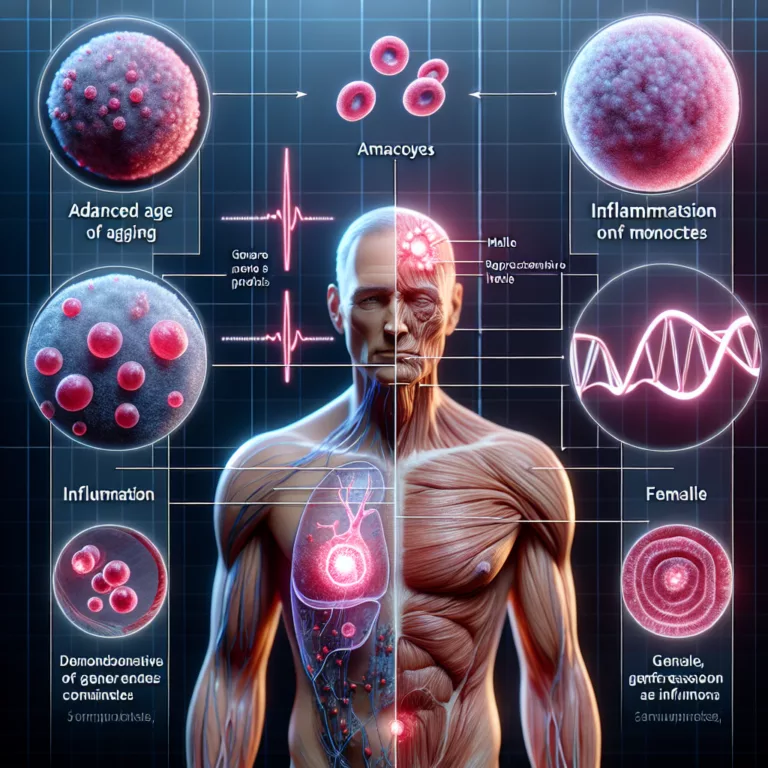
Le système immunitaire humain subit des changements significatifs avec l’âge, devenant moins efficace et plus inflammatoire. Ce déclin est complexe et implique de nombreuses populations cellulaires, chacune ayant des comportements et des profils d’expression génique distincts. L’interaction entre ces cellules et d’autres tissus ainsi que des molécules extracellulaires façonne la réponse immunitaire globale. Le vieillissement…